How to upload new Genotype data to easyGWAS? |
To perform GWAS on private data the user has the opportunity to upload new genotype, phenotype, covariate and gene annotation data to easyGWAS. All data will be integrated privately in the user's account. After the integration took place, the owner of the data can run GWAS and meta-analyses on these data. Every user has 5 Gb of easyGWAS cloud storage to integrate data.
To upload new Genotype data or whole datasets containing Genotype, Phenotype, Covariate and Gene Annotation data the user has to upload a ZIP archive with all files to easyGWAS.
Because, these ZIP archives can be rather large one cannot directly upload the data to easyGWAS via the web front end. The user needs to link easyGWAS with their own Dropbox account.
We here describe how the user can use Dropbox to upload new data to easyGWAS.
General Requirements
First the user has to prepare a ZIP archive with all the data. The archive must contain at least the genotype data in PLINK [1] format. Thus, the archive must contain a genotype.ped and a genotype.map file. Details about the exact format can be found in the following FAQ
Optionally, the archive can contain Phenotype data (phenotypes.pheno), Covariate data (covariates.cov) or a new Gene Annotation File (geneinfo.gff).
The total size for the ZIP archive should not exceed 2GB. The following table summarizes all the files and requirements for the files.
| File Type | File Extension | File Description | Required? |
|---|---|---|---|
| genotype.ped | *.ped | Genotype PED file, contains a matrix of different samples and SNPs | |
| genotype.map | *.map | Genotype MAP file, contains a list of chromosome and position information for all SNPs in the PED file | |
| phenotypes.pheno | *.pheno | Phenotype file with the phenotypic measurements for different samples. The samples have to be the same as in the MAP file. Missing phenotypic measurements must be set to nan | |
| covariates.cov | *.cov | Covariate file with measurements for different samples. The samples have to be the same as in the MAP file. Missing measurements must be donated as nan | |
| geneinfo.gff | *.gff | Gene Annotation file in GFF2 format |
After the user creates the ZIP archive, it has to be uploaded to the user's personal Dropbox account. When the upload to Dropbox is finalized, the user can start to integrate the data into easyGWAS.
How to upload new data to easyGWAS
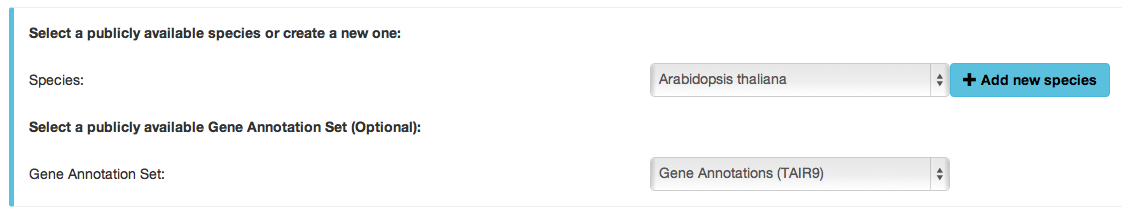
easyGWAS has already integrated different publicly available datasets for different publicly available species. The user has the option to upload a new private dataset to an existing species or to create a new private Species. Here we describe all the necessary steps to integrate private data:- Select a species of your choice or create a new one
- Select a Gene Annotation Set: Here the user can either select an available Gene Annotation Set or not. If the user decides to not select an available annotation set he/she has to possibility to link his dataset to a new private Gene Annotation Set by uploading it to easyGWAS with the new data.
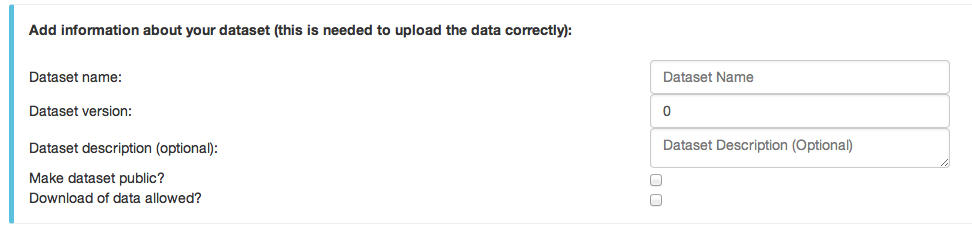
- In the next step the user must provide information about the dataset, e.g. a dataset name.
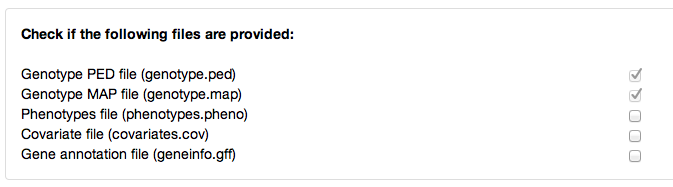
- Here the user has to specify which data to integrate into his/her easyGWAS account. Please note that the genotype data is mandatory. All other data are optional.
- In the last step the user has to select the ZIP file from his/her personal Dropbox account. For this purpose, the user has to click on "Choose from Dropbox":
Then an official and secure Dropbox popup window opens and the user has to enter his or her Dropbox credentials to link the personal Dropbox account with easyGWAS:
The users then selected his or her ZIP archive and hits the button Upload Data
Then the data is processed by the easyGWAS servers and will be integrated to the users account. The current integration status can be seen in the home view of the Upload Manager. Further the Upload Manager gives information if the data was successfully integrated and shows an error log if something went wrong.
After a successful integration the user can perform GWAS and meta-analysis with this data
References